心血管病は依然として世界における主要な死因の一つであり、新たな治療法の開発が急務である。そのためには、臨床における現象を基礎研究におけるモデルに落とし込んで、心血管病の病態生理を明らかにするとともに、そこで得られた知見を臨床応用可能な状態に変換するプラットフォームの開発が重要である。
心血管病の病態生理は、様々な細胞内シグナル伝達経路、細胞間相互作用及び多臓器連関が絡み合った複雑系により構成される。当研究室では、心血管病の病態生理のシステムとして理解を目指すとともに、システムとしての介入を目的とした研究を行っている。本項においては、我々がこれまで得た知見を示すとともに、将来の方向性について紹介する。
心疾患における血管内皮細胞に着目した細胞間相互作用
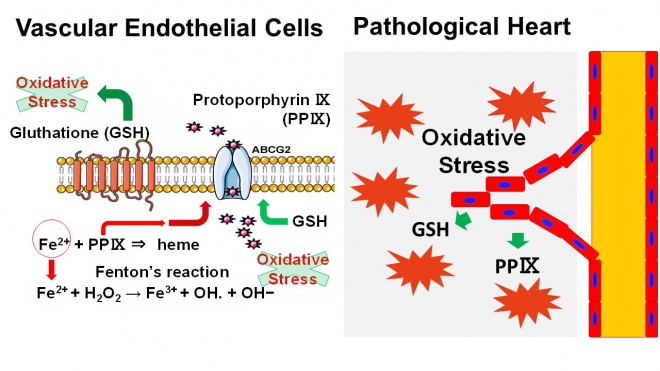
心機能の主な担い手は心筋細胞であるが、その機能は血管内皮細胞、線維芽細胞や免疫細胞等の非心筋細胞により裏打ちされている。特に血管内皮細胞は心筋細胞への血液灌流を維持するために重要な細胞であるが、我々はこの血管内皮細胞が抗酸化ストレス作用を有しており、自分自身の生存による心血流の維持と心筋細胞を主体とする心組織全体の酸化ストレス軽減による心保護作用により、病的負荷の加わった心臓における心機能維持に重要な役割を果たしていることを明らかにした(図1)。
血管内皮細胞はABCG2という膜輸送蛋白を発現している。このABCG2は心組織における他の細胞には発現していない。我々は、ABCG2欠損マウス(ABCG2KOマウス)を使用して、血管内皮細胞のABCG2を介した機能の解明を行った。
ABCG2KOマウスに前下行枝の結紮による心筋梗塞を誘導すると、野生型マウスと比較して著明に心破裂による死亡率が増加し、さらにその後の心臓の構造的・機能的変化(心リモデリング)による心拡大と心機能の低下が増悪する。心組織を解析すると、ABCG2KOマウスでは梗塞境界領域の血管新生が抑制されているとともに、組織修復が障害されていた。そのメカニズムを明らかにするため、ABCG2が細胞内から細胞外へ輸送する基質の検討を行った。その結果、ABCG2はヘムを構成するプロトポルフィリンⅨの血管内皮細胞における輸送に重要な役割を果たしていた。プロトポルフィリンⅨは細胞内の酸化ストレスの供給源となる鉄の代謝に深く関与している。以上から、血管内皮細胞はABCG2を介して細胞内酸化ストレスを軽減することにより虚血心組織における自身の生存を確保し、心組織の修復を促進していることを明らかにした。
ABCG2KOマウスに大動脈縮窄術による圧負荷心肥大を誘導すると、その後のマウスの死亡率及び心リモデリングが野生型マウスと比較して増悪した。心組織における遺伝子発現を検討すると、酸化ストレス応答系遺伝子群の発現がABCG2KOマウスで亢進していた。それに対し、酸化ストレス産生系遺伝子群の発現は野生型マウスと比較して有意な差を認めなかった。心組織全体における酸化ストレスはABCG2KOマウスで悪化しており、さらに抗酸化薬の投与によりこの表現型が改善することから、ABCG2は負荷心筋における酸化ストレスの軽減に関与し、心リモデリングを抑制することが明らかとなった。我々は、血管内皮細胞からABCG2を介して排出される物質として抗酸化物質であるグルタチオンを同定し、実際圧負荷後のABCG2KOマウスの心臓及び血漿中のグルタチオン濃度が野生型マウスと比較して低下していることを明らかとした。以上から、心筋細胞を主体とする心組織における酸化ストレスの軽減には、血管内皮細胞が重要な役割を果たしており、心肥大の病態生理に深く関与することが明らかとなった。
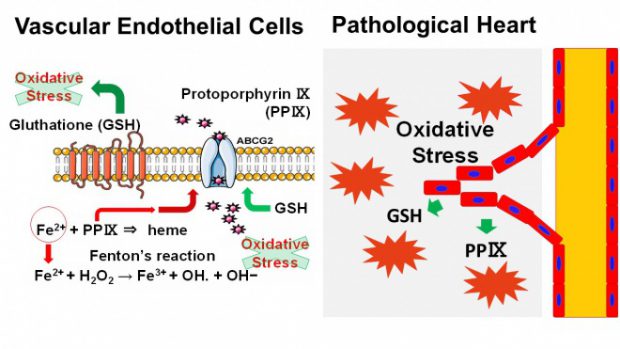
図1 心疾患における血管内皮細胞の抗酸化ストレス作用 血管内皮細胞は膜輸送蛋白ABCG2を介したプロトポルフィリンⅨ及びグルタチオンの細胞外への排出により、血管内皮細胞自身及び心組織全体の酸化ストレスを軽減する。その結果、心筋梗塞においては血管新生及び組織修復を促進し、圧負荷心肥大においては心リモデリングを抑制する。
心疾患における自然炎症の役割と臓器連関
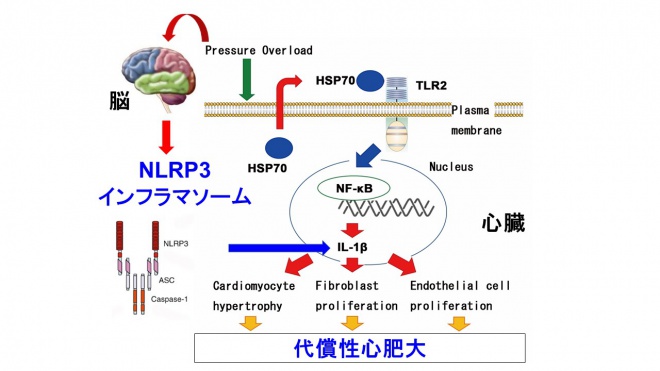
負荷が加わった心組織では炎症反応が誘導される。しかし、心組織における炎症誘導メカニズムとその制御機構は不明であった。非感染性の炎症反応は「自然炎症」とも呼ばれ、自己由来内因性リガンドと病原体センサーとの相互作用により誘導される。我々は、この自然炎症が心リモデリングに重要な役割を果たしていることを明らかとした。さらに、心臓における自然炎症が自然免疫受容体を介して誘導され、心脳連関と転写因子NF-kBの多段階制御機構により時間的空間的に制御されていることを明らかにした(図2及び図3)。
自然免疫受容体TLR2は心組織を構成する細胞に広く発現している。このTLR2を欠損したマウス(TLR2KOマウス)に圧負荷心肥大を誘導すると、代償性心肥大が障害され、心機能が低下することが分かった。野生型マウスの骨髄をTLR2KOマウスに移植しても表現型は変化を認めなかったことから、非免疫細胞の重要性が明らかとなった。心組織を解析すると、TLR2の下流にある転写因子NF-kBの活性化及び炎症性サイトカインIL-1βの発現増加がTLR2KOマウスで抑制されていた。実際、野生型マウスにおけるNF-kB活性及びIL-1βの抑制はTLR2KOマウスと同様の表現型を示した。細胞レベルで解析をすると、TLR2/NF-kB/IL-1β系は心筋細胞肥大、線維芽細胞増殖及び血管内皮細胞増殖を制御していた。我々は、TLR2/NF-kB/IL-1β系を活性化させる物質として、熱ショック蛋白質Hsp70を同定した。Hsp70は主に心筋細胞から分泌されており、圧負荷後の血漿中の濃度が増加する。我々は、このHsp70が実際にin vitro及びin vivoでTLR2/NF-kB/IL-1β系を活性化させ、代償性心肥大に重要な役割を果たしていることを確認した。以上から、心組織における炎症反応は、autocrine及びparacrineのメカニズムによるTLR2/NF-kB/IL-1β系の活性化により誘導され、圧負荷時の心リモデリングに重要な役割を果たすことを明らかにした。
IL-1βの活性化には自然免疫受容体NLRP3インフラマソームの活性化が重要である。実際、野生型マウスに圧負荷心肥大を誘導すると、心組織におけるNLRP3インフラマソームは活性化される。NLRP3欠損マウス(NLRP3KOマウス)に圧負荷を加えると、TLR2KOマウスと同様の表現型を示した。我々は、圧負荷時のNLRP3インフラマソームの活性化機構として、細胞外ATPによるプリン作動性受容体P2X7の活性化が重要であることを明らかにした。実際、薬物による細胞外ATP除去処理をした野生型マウスとP2X7欠損マウスに圧負荷を加えると、圧負荷後心組織におけるNLRP3インフラマソームの活性化及び代償性心肥大が抑制された。さらに我々は、ATPバイオセンサーシステムを用いて、圧負荷後の心組織における細胞外ATP濃度の計測を行った。その結果、圧負荷後の心組織では実際に細胞外ATP濃度が増加しており、各種モデルを使用した検討から、その供給源は交感神経終末であることを明らかにした。また、我々は交感神経終末からのATPの分泌には、心臓から脳への求心性神経線維の活性化が必要であることを明らかにした。以上から、心臓における炎症誘導シグナルは心脳連関により制御されており、恒常性維持に重要な役割を果たすことが分かった。
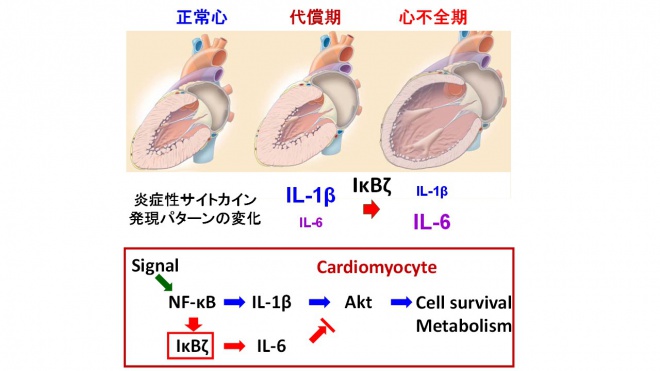
我々は自然炎症による心臓恒常性維持機構の重要性を明らかとしたが、一方で炎症反応は心不全を引き起こす。そこで我々は、代償期及び心不全期の炎症性サイトカインの発現パターンの違いに着目して、自然炎症が心臓の恒常性維持機構の破綻をもたらすメカニズムの解明を試みた。圧負荷を加えた野生型マウスでは、代償期にはIL-1βの発現が、心不全期にはIL-6の発現が優位となる。我々は、NF-κBシステムにおいてIL-1β優位からIL-6優位への発現パターンの変化に関与している転写制御因子IκBζに着目した。実際、IκBζの発現は代償期と比較して心不全期に有意に増加している。そこで、IκBζのヘテロノックアウトマウスに圧負荷を加え、心臓の表現型を評価したところ、代償性心肥大は誘導されたが、心不全への移行は認めなかった。心組織における炎症性サイトカインの発現パターンはIL-1β優位の発現パターンであった。また、我々は、このIL-1β優位の発現パターンがAktの活性化に重要であることを明らかにした。以上から、心組織における炎症反応の時間的変化はIκBζにより制御されており、心臓の恒常性維持機構の破綻に重要な役割を果たすことが分かった。
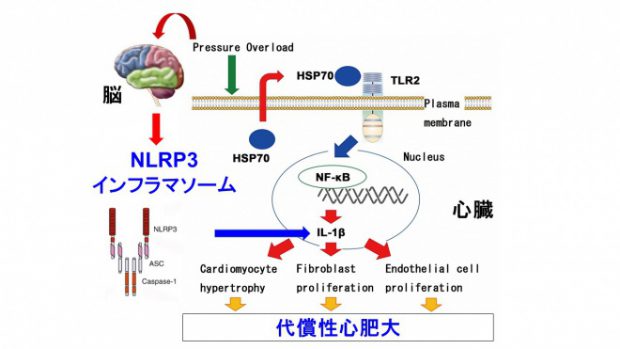
図2 心疾患における自然炎症の空間的制御機構 心疾患の病態生理においては、自然炎症が重要な役割を果たす。心臓における自然炎症は、autocrine及びparacrineのメカニズムで自然免疫受容体TLR2の活性化を介して誘導され、代償性肥大に関与する。さらにTLR2を介した炎症性サイトカインIL-1βの産生は心脳連関によるNLRP3インフラマソームにより制御される。
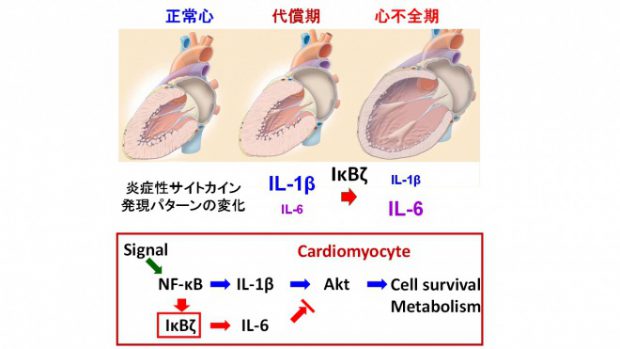
図3 心疾患における自然炎症の時間的制御機構 心臓に病的負荷が加わると、早期においては代償性肥大が誘導されるが、時間の経過とともに心不全へと移行する。自然炎症は代償性肥大及び心不全への移行の両者に関与する。この時間的変化は転写制御因子IκBζによる炎症性サイトカインの発現パターンの制御機構により説明される。
動脈硬化病変における血管外膜微小血管の役割
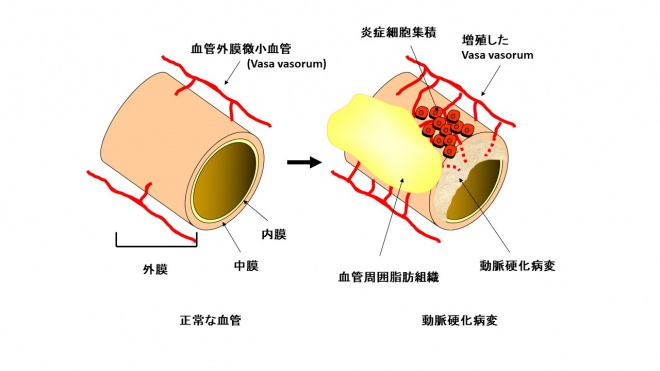
動脈硬化病変研究では主に血管壁細胞やプラーク構成成分が注目されてきたが、一方で、発達した動脈硬化病変では外膜に微小血管(vasa vasorum, VV)増殖を認めることも古くから報告されている(図4)。
しかし、VV増殖は、動脈硬化病変進展の結果として生じるのか、VV増殖が病変を進展させるのか、明らかになっていない。我々は、動脈硬化モデルマウスであるApolipoprotein E (ApoE)欠損マウスを用いて血管外膜VVの役割を検討した。高齢ApoE欠損マウスの腹部大動脈に自然発生した動脈硬化病変の外膜では、病変拡大に伴いVV増殖を認めた。次に、病変を持たない若年ApoE欠損マウスの腹部大動脈周囲に、徐放化した塩基性線維芽細胞増殖因子(bFGF)を留置し血管外膜VV増殖を強制的に生じさせたところ、外膜への炎症細胞集積および動脈硬化病変の形成と拡大を認めた。以上より、外膜VV増殖の調節により動脈硬化病変の進展にも影響を及ぼす可能性があると考えられた。現在、様々な血管病変モデルでの外膜VV増殖形態の差異や、外膜VVを標的とした動脈硬化病変治療の可能性について検討中である。
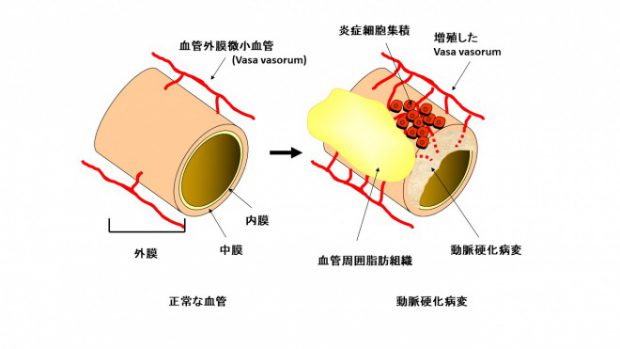
図4 動脈硬化病変における血管外膜微小血管増殖 正常な血管では外膜微小血管(vasa vasorum)は血管外膜側の栄養血管であるが、拡大した動脈硬化病変では増殖して外膜側から動脈硬化病変内に侵入し、病変内に脂質や炎症細胞を送り込む導管となるといわれている。
システムとしての介入を目指した新規治療プラットフォームの開発
心血管病の病態生理は、時間及び空間的変化を支配する複数のシステムの全体的な偏移、セットポイント・シフトとしての側面が強いと考えられる。よって、単一の要因に対する無秩序なシステムへの介入では十分な治療効果を得ることは難しいことが予想され、システムに対しシステムで制御する方法の開発が必要である。我々は、合成生物学的手法を用いて、時間空間分解能をもった次世代治療プラットフォームの開発を目指している。
細胞のふるまいを制御するためには、memory systemとlogic operation systemの開発が必要である。我々は、CRISPR/Cas9システムを使用して、pseudorandom memory systemとdirectional memory systemの両者の開発を行っている他、合成遺伝子回路を使用したlogic operation systemの開発を行っている。さらに、directional memory systemはlogic operation systemとの連結が可能であり、過去のbiological informationを未来の細胞のふるまいの制御に反映させることが可能となりうる。現在はまだproof-of-conceptの段階であるが、この分野の技術開発は爆発的なスピードで進んでおり、近い将来、臨床応用可能な技術となりうると考えられる。